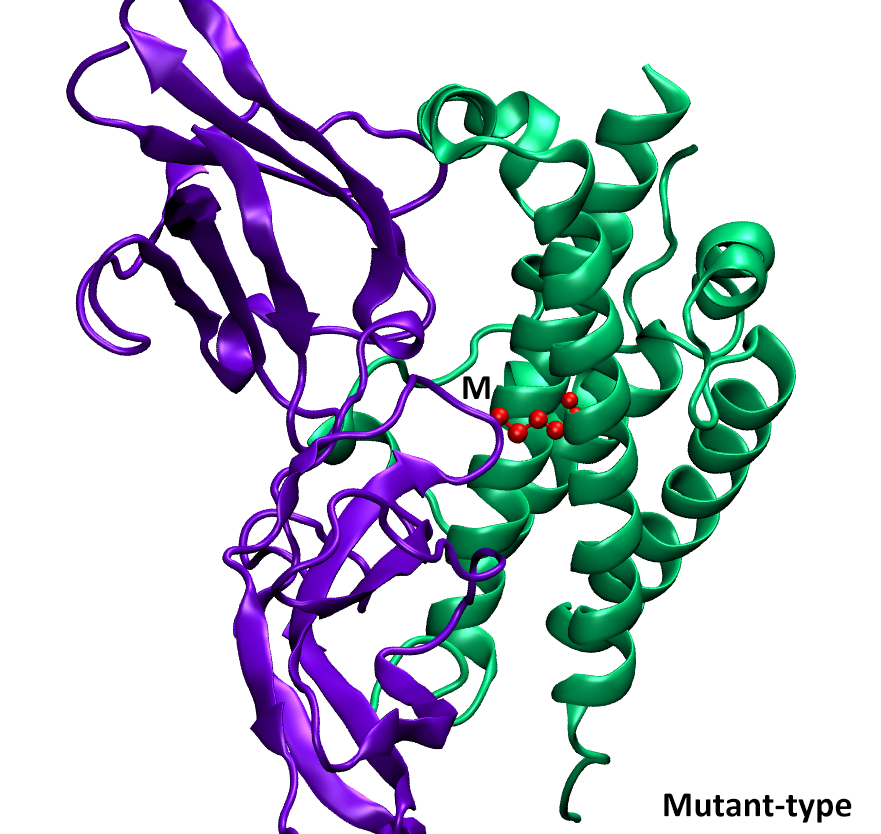
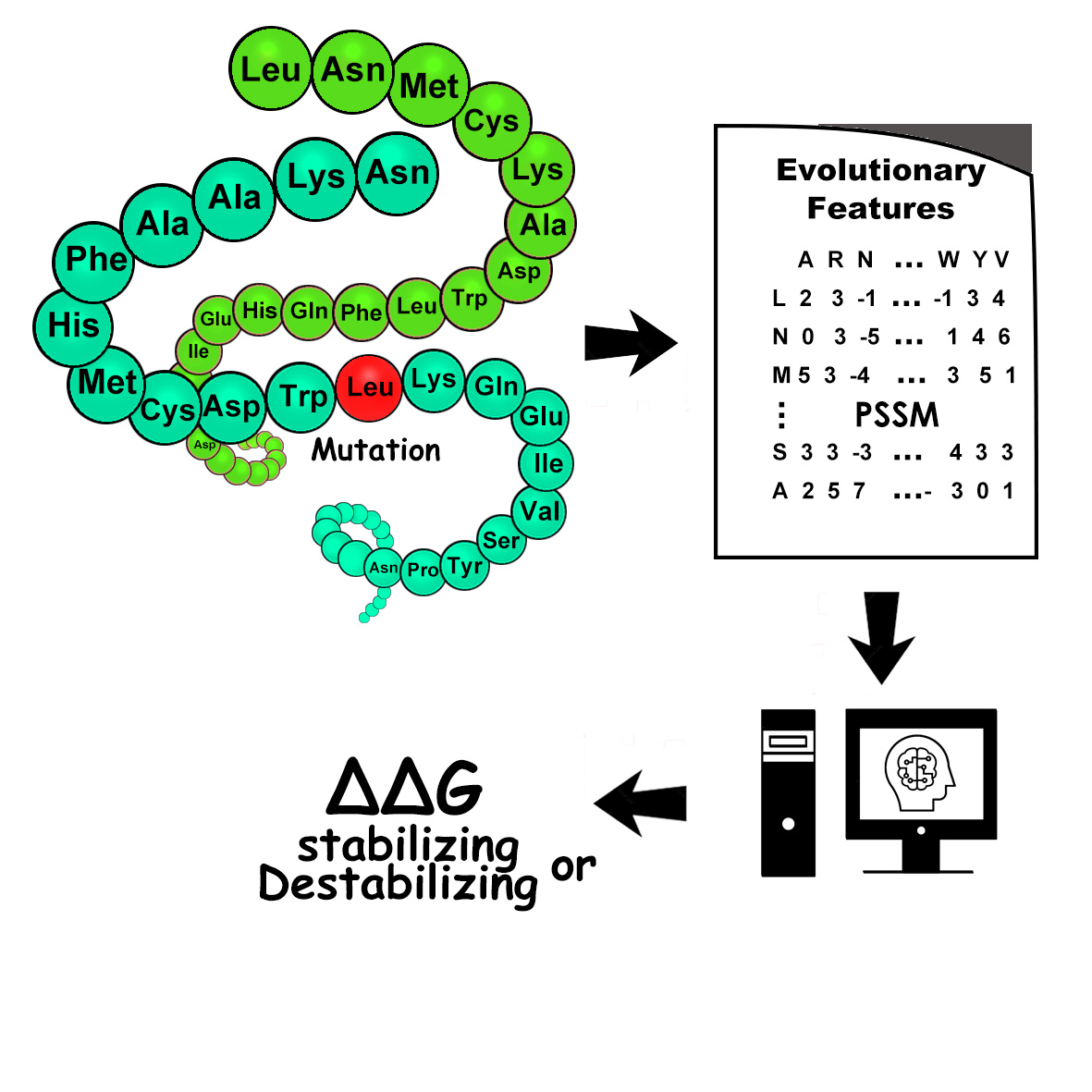
SAAMBE is curently processing your file, check back later for updates.

Single Amino Acid Mutation change of Binding Energy (SAAMBE) method provides several algorithms and webservers to assess the effects of mutations on protein-protein interactions (PPIs). To address different types of investigations, here we provide several versions of SAAMBE, listed below.
17,119 Total views.